



Sickle cell disease is seen increasing rapidly globally, it is a disease affecting the blood. Due to this disease, the level of hemoglobin within the blood cells starts getting affected. For more information watch the video till the end.
Sickle cell disease (SCD) is a serious inherited blood disorder that impacts millions of people worldwide. While its presence is being increasingly recognized, SCD is not a new condition. This comprehensive overview dives into the complexities of SCD, exploring its causes, symptoms, diagnosis, treatment options, and the ongoing fight to improve the lives of those affected.
Understanding Hemoglobin and Red Blood Cells
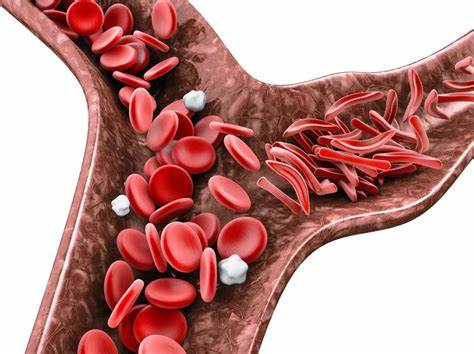
Before delving into SCD, it’s crucial to understand the role of hemoglobin and red blood cells. Hemoglobin is a protein present within red blood cells, responsible for carrying oxygen throughout the body. Healthy red blood cells are disc-shaped and flexible, allowing them to effortlessly navigate even the narrowest blood vessels and deliver oxygen to every tissue.
The Disruption: Sickle Hemoglobin and its Consequences
In SCD, a genetic mutation alters the structure of hemoglobin, causing it to become sickle-shaped under certain conditions. These sickle-shaped red blood cells are rigid and sticky, leading to a cascade of problems:
- Blocked Blood Flow: Sickle cells clump together, obstructing blood flow to various organs and tissues. This blockage restricts oxygen delivery, causing organ damage and immense pain, a hallmark symptom of SCD.
- Reduced Oxygen Delivery: The abnormal shape of sickle cells also hinders their ability to efficiently carry oxygen. This oxygen deficiency, termed anemia, further contributes to fatigue, weakness, and other complications.
- Increased Vulnerability to Infections: The impaired function and lifespan of sickle cells weaken the body’s defense system, making individuals with SCD more susceptible to infections.
The Spectrum of Sickle Cell Disease
SCD is an umbrella term encompassing various subtypes, each with slightly different characteristics and severity. The most common form is sickle cell anemia, but other variants include:
- Sickle-hemoglobin C disease
- Sickle beta-thalassemia
The severity of symptoms and potential complications can vary depending on the specific subtype a person has.
Unveiling the Cause: Genetics and Inheritance
Sickle cell disease is an autosomal recessive genetic disorder. This means that an individual inherits two abnormal copies of the gene that controls hemoglobin production, one from each parent. If a person inherits only one abnormal copy (trait carrier), they typically won’t develop SCD but can pass the trait on to their offspring.
The prevalence of SCD is highest in populations with a history of malaria, as the abnormal hemoglobin gene offered some protection against this deadly parasite. However, this comes at the cost of increased susceptibility to SCD if both parents carry the trait.
Recognizing the Signs and Symptoms
The symptoms of SCD can manifest at a young age, typically around 5-6 months. Common indicators include:
- Severe pain episodes (vaso-occlusive crisis) in various parts of the body, often triggered by infection, dehydration, or stress.
- Fatigue and weakness due to anemia
- Frequent infections
- Delayed growth and development in children
- Swelling in hands and feet
- Yellowing of the eyes and skin (jaundice)
The severity and frequency of these symptoms can vary significantly between individuals.
Diagnosis: Confirming the Presence of SCD
A blood test is the standard method for diagnosing SCD. This test analyzes the structure and function of hemoglobin to identify abnormalities. Newborn screening programs in many countries help ensure early detection and intervention.
Living with SCD: Management and Treatment
While there is no cure for SCD, a range of treatment options can help manage the condition and improve quality of life. These include:
- Pain Management: Medications like pain relievers and opioids are crucial during pain crises.
- Hydration: Maintaining adequate hydration helps prevent blood cells from sickling.
- Hydroxyurea: This medication can increase the production of fetal hemoglobin, a type that is more flexible and less likely to sickle.
- Blood Transfusions: Regular blood transfusions can replace diseased red blood cells with healthy ones.
- Bone Marrow Transplant: In severe cases, a bone marrow transplant can potentially cure SCD by introducing healthy stem cells that produce normal hemoglobin.
The Future of SCD: Research and Hope
Researchers are actively exploring new avenues for SCD treatment and even potential cures. Some promising areas include:
- Gene Therapy: This approach aims to correct the underlying genetic mutation responsible for SCD.
- Stem Cell Therapy: Stem cell transplants offer a potential cure, but challenges related to donor matching and transplant risks remain.
- New Medications: Development of drugs that target the sickling process or enhance the production of healthy hemoglobin is ongoing.
Increased public awareness, improved screening programs, and ongoing research offer hope for a future where SCD’s impact is significantly diminished.